
Arithmetic Outsmarts Seven Deep Neural Networks
Simple sums humble single-cell foundation models in CRISPR prediction

I just finished reading the new Nature Methods study (Deep-learning-based gene perturbation effect prediction does not yet outperform simple linear baselines) that pits arithmetic against neural heft in single-cell CRISPR prediction. The authors pulled four open datasets covering 224 double edits and hundreds of single-gene perturbations. Into that arena they dropped seven networks: scGPT, scFoundation, scBERT, Geneformer, UCE, GEARS, and CPA. Two baselines kept them honest: a no-change guess and a plain additive rule.
The additive rule keeps both feet on raw data. For a double hit on genes A and B it forecasts
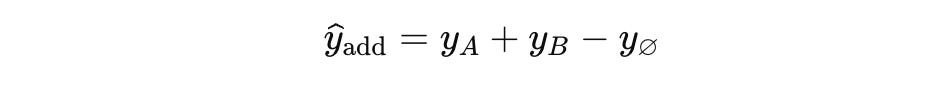
where yA and yB are the measured single-edit shifts and y∅ is the unedited control. No weights, no tuning.
Every model receives the same training menu: all 100 single-gene profiles and roughly half of the double-gene cases, with five random splits holding out the rest for evaluation. That symmetry removes any information edge. Both the additive sum and the neural stacks carry the same batch effects and library quirks.
Results land hard on the side of simplicity. On the double-edit benchmark every deep network shows a larger L2 error than the additive sum. None beats even the do-nothing guess at flagging real genetic interactions, and most predictions drift toward bland buffering instead of spotting synergy or opposition.

Switch to single unseen edits in Replogle’s K562 and RPE1 screens plus the Adamson set, and nothing changes. Copying the per-gene mean or fitting a small rank-restricted linear regressor matches or outperforms every foundation model.
A compact linear model that reuses embeddings learned from a related dataset quietly outruns all seven giants. Focused, task-specific pre-training matters more than broad atlas-scale diets when examples are scarce.
Ok, Why The Gap?
Arithmetic lifts empirical vectors straight into the forecast and never risks overfitting. Meanwhile, each neural system must compress those same vectors into a latent space, merge two embeddings, and reconstruct an entire transcriptome shift. That is a tall order with only a few hundred double edits. UCE even slips a sliver of ground truth into its embedding step by overwriting relevant gene rows with observed values, yet addition still holds its own.
Transparent linear baselines now stand as the reference. Any future model claiming progress in single-cell perturbation prediction needs to clear that bar first.
I love this baseline. It has been my long standing pet-peeve about ‘GPT’ like models trying to predict biological conditions.