
A Checklist Before Geometry
A manifesto for mixed-dimensional models

If the goal is a synthetic cell that does not lie, we have to make three commitments up front:
pick the space that holds the state,
pick the operators that evolve it, and
publish the error we are prepared to carry.
Everything else is detail. But, the details decide whether the thing composes, calibrates, and controls.

At CELL, this ‘detail’ is the checklist we need to first publish and agree upon, scrutinize, and pin down. The checklist forces us to name what exists, what moves, what is measured, and what we will treat as error. Only then do we decide where the objects live and which operators are admissible.
Note: While this article is written to mathematicians, mathematical biology and biophysics audience, it is truly the empirical biologists who can keep the checklist real and provable.
The checklist helps us hypothesize what the host geometry of the mathematical configuration should possibly be.
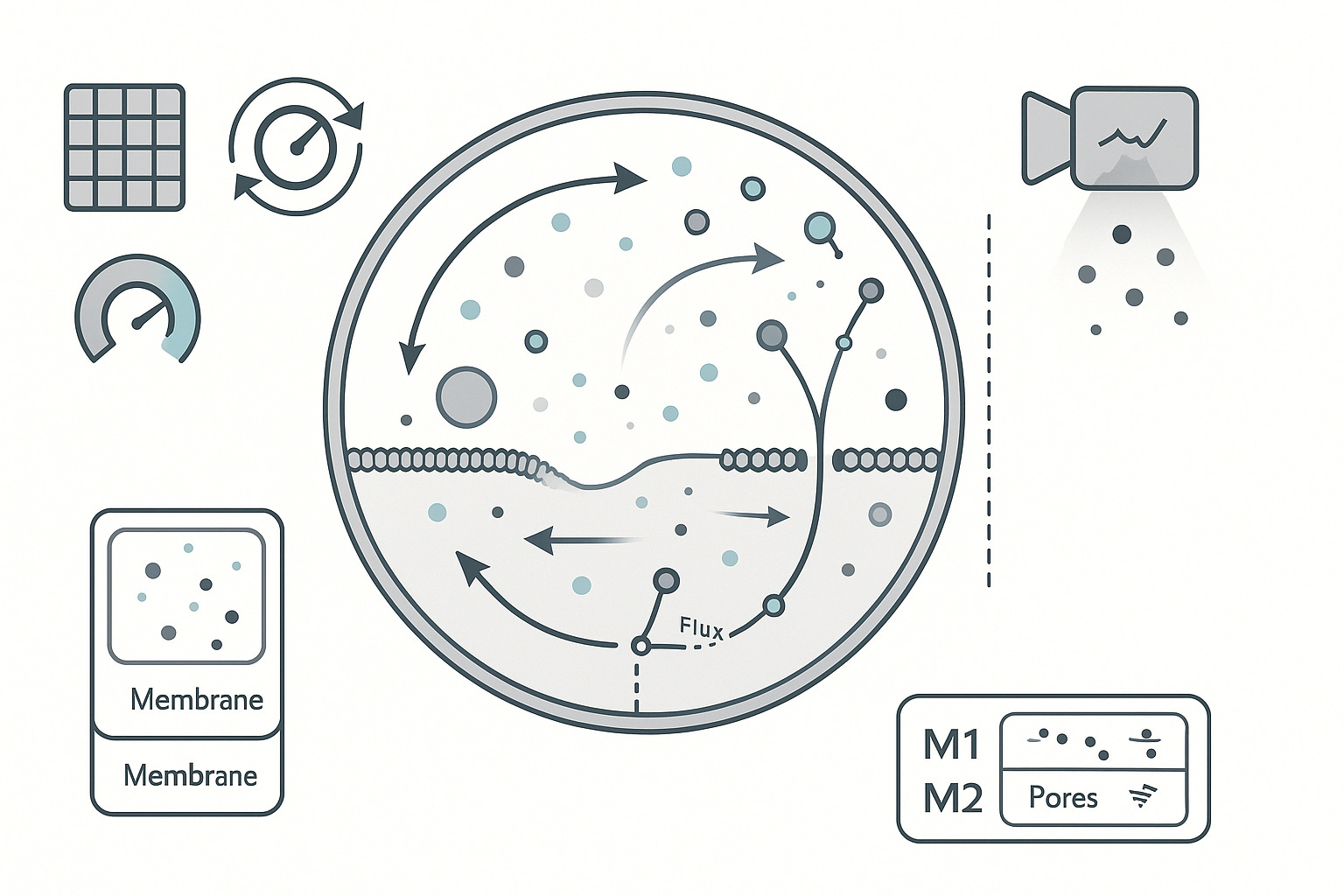
Why name a geometry at all?
We start with the checklist because it anchors what exists, what moves, and what gets measured. Only then does a geometry become more than taste. Once conservation and coupling are explicit, the host space is a hypothesis we can test, not a preference we defend. H1 is that testable proposal
One such hypothesis is as follows:
The hypothesis, stated cleanly
Hypothesis H1. The correct host geometry (or a universal configuration space) for molecular-scale models is mixed-dimensional. Volumes carry concentrations and fields, surfaces carry lateral transport and storage, thin pores and filaments carry constrained flux, and a small set of points carry discrete internal states and gates. Interfaces are first-class objects with traces and jumps, not numerical afterthoughts.
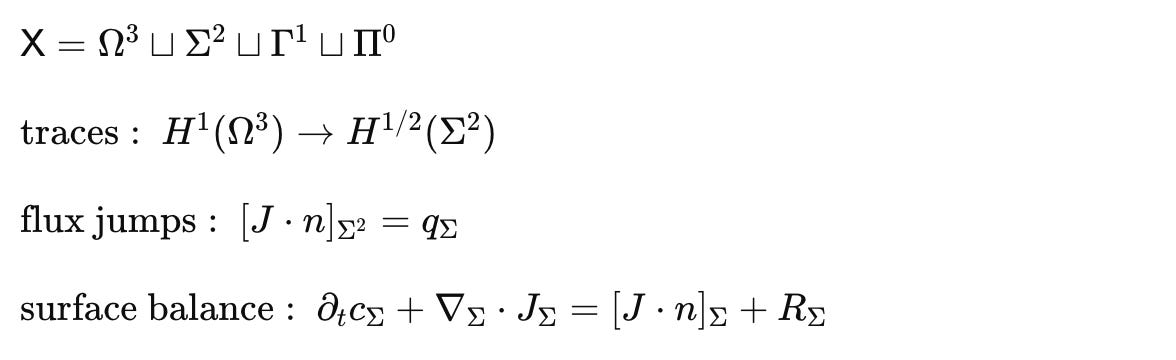
Avoiding the parameterization chasm
Progressive enrichment should be the plan. We should begin with a minimal working model that respects conservation and separates physics from measurement, then add structure only when data and falsification demand it. Promotion is earned, not assumed. For example:
M0 captures bulk concentrations, a single membrane surface with capacitance, and a clean observation model.
M1 adds boundary transport operators, surface reaction–diffusion, and heterogeneity priors with shared hyperparameters.
M2 introduces line features for pores and filaments, rare event channels, and validated cross-scale hooks.
Promotion criteria: a measurable gap in predictive skill or uncertainty coverage at fixed ε; a parameter that is identifiable under current data; a conserved quantity that would be violated without the added structure.
Why H1 is reasonable
Start with what we want to conserve and control. Mass, charge, and free energy move through membranes, pores, and scaffolds. Real cells store charge on bilayers, hold binding capacity on surfaces, and route flux through narrow structures that act as valves. If we smear those interfaces into the volume, we lose storage, delay, and selectivity. The numerical scheme then fakes them with ad hoc terms and hidden nonlocal couplings. Inference picks up the slack by distorting parameters to fit a confounded observation model.
Observable consequences if H1 is right
Electrodiffusion with classical membrane capacitance requires explicit surface storage to reproduce correct charging transients without nonlocal hacks.
Transporters and channels are identifiable only when boundary traces and partition coefficients are modeled as operators, not as lumped scalars.
Reaction and diffusion on membranes form lateral waves and clustering that cannot be reproduced by homogenizing the membrane into the volume at experimental spatial resolution.
Observation models that localize to surfaces or small structures, for example FRET near membranes or smFISH at nuclear speckles, demand surface and point-localized operators to be statistically well-posed.
How H1 can be falsified
A single volumetric model with no explicit surfaces or lines reproduces capacitance transients, channel gating kinetics, and surface diffusion at experimental resolutions with a single set of parameters across contexts.
Parameter posteriors from models with and without explicit interfaces coincide within declared error for the same datasets and priors.
Controlled interventions that act only at interfaces, for example leaflets or nuclear pores, have indistinguishable effects in models that lack those interfaces.
Minimal math to make H1 operational

Each term acts on the appropriate stratum, with coupling through traces, jumps, and partition maps that preserve conservation laws
With that in place, we can say exactly what must be captured before we argue about ambient spaces, regularity, or solvers.
Ok, what is the narrative here?
We work on mixed-dimensional domains because biology forces it. Membranes are surfaces. Pores and channels are curves. Condensates and cytosol are volumes. Interfaces are not decoration, they are the places where flux is constrained, energy is exchanged, and measurements get distorted. If we do not specify where each field lives and what traces exist on each interface, we bake non-physical leaks into the model and never notice until inference fails.
We separate physics from measurement. States and fluxes belong to the system. Photons, barcodes, peptides, and capture efficiencies belong to the apparatus. That boundary must be explicit, otherwise the learning algorithm pushes physical nonsense into the parameters to fit a confounded observation model.
We keep thermodynamics in the loop. Feasible flux directions, cycle affinities, bounded capacities, and positivity are not stylistic choices, they are the only way to guarantee that numerics do not invent energy or mass. The operator library is constrained by these invariants. If a candidate model violates them, it is rejected, no matter how well it fits a dataset.
We embrace heterogeneity as structure, not noise. Cells carry distributions over parameters and states. That is a feature to be modeled, not a nuisance to be averaged away. Hierarchical priors and random fields are not afterthoughts, they are part of the definition of S.
We publish ε because control without error budgets is theater. Calibration plans, per-observable tolerances, and prospective validation tasks tie ε to decisions. Robustness is then defined against declared inadequacy, not a fiction of perfection.
With that spine in place, here is the capture list we hold ourselves to at the molecular scale. This is what we collect before we argue about the best ambient space or solver.
What does a list look like, you ask. Here is a first cut:
Universal capture list, molecular scale
Physical domain and geometry: compartment atlas and subcompartment meshes for eukaryotic cells, organelles, membranes (both leaflets), pores and channels, phase-separated condensates, microdomains, extracellular peri-cellular space; interface topology and boundary types, surface curvatures, and contact sets between compartments.
Species catalog and ontologies: all molecular species and complexes with internal state labels: genes, transcripts, splice isoforms, proteins with post-translational modifications, metabolites, lipids, ions, cofactors, RNAs with RBP occupancy, chromatin states, motor proteins, structural polymers, scaffolded assemblies.
State variables per species:
• Copy number or concentration fields in space and time
• Discrete internal states (conformation, binding, phosphorylation count, nucleotide state)
• Localization state across compartments and microdomains
• For polymers: contour configurations, loop states, tethering and anchoringReaction network structure: stoichiometric matrix S, reaction channels, reversibility flags, catalysts, inhibitors, conserved moieties, deficiency, linkage classes, elementary steps versus coarse reactions, mechanochemical and electrodiffusive couplings.
Kinetic laws and timescales: rate laws per reaction (mass action, Michaelis–Menten, Hill, microkinetic schemes), allosteric modulation, saturation and crowding corrections, detailed balance constraints for equilibrium subcircuits, separation into fast and slow manifolds with quasi-steady reductions.
Transport processes: diffusion tensors per species and region, advection fields, active transport processes, electrophoretic drift, membrane permeabilities, facilitated transporters, pumps, channel gating states, nucleocytoplasmic transport, vesicular trafficking kernels.
Electrodiffusion and electrostatics: local charge densities, ionic strength, dielectric properties, Debye lengths, electrostatic potential and its coupling to fluxes, boundary capacitances at membranes, field-dependent reaction and transport corrections.
Thermodynamics and energetics: standard chemical potentials, Gibbs free energies and energy landscapes for conformational changes, ATP and redox budgets, heat production, entropy production rates, nonequilibrium driving forces, cycle affinities, thermodynamic feasibility of fluxes.
Stochastic structure: intrinsic reaction noise (jump processes), diffusive noise for mesoscopic limits, extrinsic fluctuations, noise covariance structure, rare event channels and large-deviation rates, switching times between metastable states.
Molecular mechanics and structure: coarse-grained coordinates for macromolecules, contact graphs, binding site geometries, elastic moduli for polymers, chromatin fiber models, protein folding basins, phase-separation order parameters and exchange rates.
Interfaces and boundary conditions: Dirichlet, Neumann, Robin, and mixed conditions per species and interface; flux continuity with partition coefficients; curvature-coupled reaction modifiers on membranes and condensate boundaries.
External inputs and actuators: ligand dosing fields, optogenetic light fields, temperature and pH profiles, osmolarity, mechanical stress at molecular attachment points, CRISPR editing events, drug schedules, ionophore actions, media composition changes.
Observation and measurement model: mapping from hidden states to observables for all modalities: fluorescence with point spread function and shot noise, FRET efficiencies, smFISH counts, RNA-seq sampling and capture efficiencies, mass-spec peptide detectability, ChIP-seq and ATAC-seq biases, calibration constants and batch effects.
Constraints and invariants: mass and charge conservation, positivity, unit consistency, stoichiometric compatibility classes, thermodynamic consistency, bounded capacities for binding sites and transporters, hard saturation constraints.
Heterogeneity and priors: cell-to-cell parameter distributions, allele-specific effects, isoform usage distributions, epigenetic heterogeneity, prior distributions on kinetic parameters and transport coefficients; correlations across parameters induced by biophysics.
Environment and solvent: viscosity and macromolecular crowding fields, excluded-volume fractions, effective diffusion corrections, ionic composition, buffer systems, local dielectric heterogeneity, hydration layers.
Redox and metal cofactors: NADH and NADPH pools, glutathione redox couple, oxygen tension, metal ion availability and buffering, metalloprotein occupancy.
Genome context and regulation: DNA sequence and accessibility, promoter and enhancer maps with binding energy models, chromatin states and 3D genome architecture, topologically associating domains, transcription factor occupancy dynamics, promoter proximal pausing.
RNA life cycle: transcription initiation, elongation, termination, splicing choices, editing, nuclear export, localization, decay pathways, RBP binding kinetics, microRNA interactions.
Protein life cycle: translation initiation and elongation, co- and post-translational modifications, folding and chaperone interactions, trafficking and sorting signals, complex assembly, degradation pathways and ubiquitin dynamics.
Metabolism: full stoichiometric matrix for metabolites, enzyme kinetics, thermodynamic constraints on flux directions, cofactor cycles, energy coupling, exchange with extracellular space, constraint-based limits alongside kinetic models.
Signaling cascades: receptor–ligand binding and proofreading, adaptor and scaffold occupancy, kinase and phosphatase networks, second messenger dynamics, crosstalk maps, feedback and feedforward motifs with delays.
Molecular assemblies and condensates: scaffold–client rules, saturation thresholds, rheology of condensates, selective partitioning coefficients, nucleation rates, aging and material state transitions.
Cytoskeletal and motor coupling: polymerization dynamics, filament aging, catastrophe and rescue, motor stepping rates and load dependence, clutch models at adhesion points, mechanochemical feedbacks.
Boundary microchemistry of membranes: lipid composition fields, leaflet asymmetry, flip-flop dynamics, raft domains, curvature composition coupling, membrane tension fields.
Time bases and clocks: absolute and relative timescales, circadian and ultradian drives, cell-cycle phase, synchronization sources, experiment time stamps and alignment rules.
Cross-scale hooks: fluxes and traces to the cellular-scale state variables, coarse observables and order parameters, upscaling operators and homogenized coefficients to bridge to tissue models.
Operator library: diffusion, advection, reaction, electrodiffusion generators, nonlocal kernels for long-range interactions, fractional operators where appropriate, plus their discrete counterparts for network subspaces.
Regularity classes and domains for variables: target regularity per field to ensure well-posedness and numerics, admissible function spaces for potentials, fluxes, concentrations, measures for low copy numbers, and traces on interfaces (to be finalized after we pick the space).
Error budget ε and validation targets: per-observable tolerances, model inadequacy terms, calibration plan, cross-validation splits aligned to modalities, prospective checks in the Validation Arena.
Provenance and units: coordinate frames, reference conditions, unit registry, versioned metadata, sheaf-like restriction maps for open sets of the domain so that pullbacks and traces are well defined across subregions.
You might be thinking. “Phew, what the hell bruh…, so, you are not asking for a checklist, but the entire universe?”.
Ahem, yes and no. Allow me to clarify. It looks like the universe because it is the superset we should use to keep submodels honest and composable, not a demand to parameterize everything today.
Think of it this way: the “universal” list is a (organic, growing) registry of variables, interfaces, units, and invariants so that any minimal model can say exactly what it includes, what it ignores, and which conservation laws still hold. We should work by progressive enrichment: start with a small, runnable M0 core, then promote only the features that close measurable gaps at a fixed ε. No single team fills the table. Each contributor ships a narrow Capture Pack tied to a context, with data and code, so the field advances in bounded steps while staying compatible with a larger geometry.
Why this matters
Geometry is not aesthetics. It is bookkeeping for conservation and coupling.
Measurement is not a footnote. It is the only way inference stays honest.
ε is not an afterthought. It is the contract that turns a model into a decision tool
If H1 is right, interfaces are not decoration. They are where mass, charge, and information are stored, exchanged, and delayed. If H1 is wrong, we should be able to show a single bulk model that transfers across contexts without retuning. Either way, we win knowledge.
Open call
Pressure test H1, or replace it. Bring the cleanest confirmation or the sharpest counterexample. We must come together to build and publish runnable artifacts, not a claim.
Plus: units, coordinate frames, observation model, priors, code, data
If your result holds across contexts without retuning, say so. If it fails, say where and why. The field moves on the strength of reproducible failures and durable wins.
Join us to debate what the configuration space should look like. What should be the correct hypothesis. What are the correct level of details to capture.
We are looking for advisors to keep us honest and grounded. We are looking for contributors to move the needle of science forward in spirit of open science. This is the only way forward.
Minimal ask for now: share this with one person in the domain. Subscribe to the Substack and join the channel. This is where all the grass-root efforts start, and you are part of it.
Subscribe: https://cellbiosf.substack.com
Join our channel: https://x.com/cellbiosf